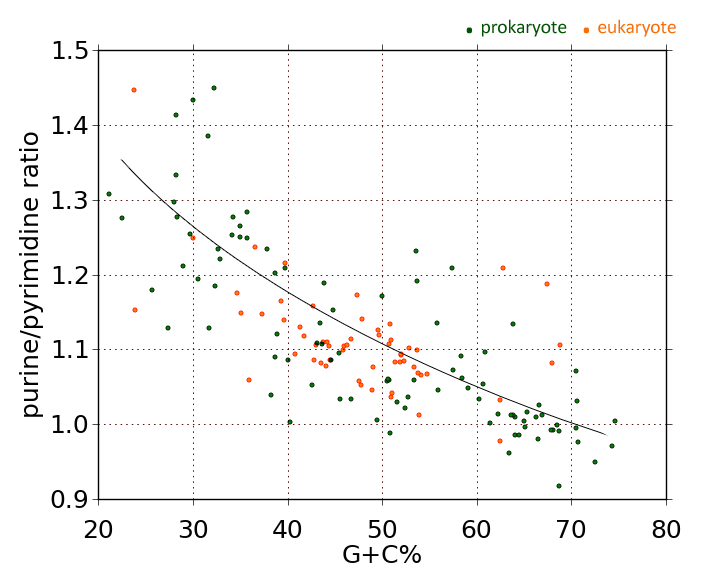
To recap my desktop-science experiments of the last month or so, I've found strandwise DNA asymmetry across domains, which is to say in bacteria, Archaea, eukaryotes, viruses, and mitochondrial DNA. In every case except mitochondria, the message (or RNA-synonymous) strand of DNA in coding regions tends to be purine-rich. The opposite strand tends to be pyrimidine-rich. Moreover, in all domains, including mitochondria, message-strand purine content increases in proportion to genome A+T content. (A+T content is a phylogenetic signature. Some genomes are inherently high in A+T content—or low in G+C content—while others are not. Related organisms tend to have similar A+T or G+C contents.)
Mitochondrial genes tend to be pyrmidine-rich on the message strand, seemingly in violation of the finding that in all other domains, message strands are purine-rich. The mitochondrial anomaly is actually very easy to understand (although it took me weeks to realize the explanation). In a nutshell: Mitochondrial DNA is pyrimidine-rich on message strands because mtDNA encodes only a few proteins (13, usually), all of them membrane-associated. Membrane-associated proteins are unusual because they tend to incorporate mostly non-polar amino acids such as leucine, isoleucine, valine, proline, alanine, or phenylalanine—all of which are specified by pyrimidine-rich codons.
It seems to me mitochondrial DNA shouldn't be thought of as a genome, because well over 90% of mitochondrial-associated gene products are encoded by genes in the host nucleus. (In humans, there may be as many as 1500 nuclear-encoded mitochondrial genes.) This point is worth repeating, so let me quote Patrick Chinnery, TRENDS in Genetics (2003) 19:2, 60:
The circular mitochondrial "chromosome" (if it can be called that) is the vestigial remnant of a much larger genome that long ago migrated to the host nucleus, no doubt to avoid oxidative attack. The mitochondrion simply is not a safe place to store DNA. (Would you set up a sperm bank in a rocket-fuel factory?) It's teeming with molecular oxygen, superoxides, peroxides, free protons, and other hazardous materials.
Human mitochondrial DNA (which is typical of a lot of mtDNA) encodes just a handful of multi-subnit transmembrane proteins, namely: cytochrome-c oxidase, NADH dehydrogenase, cytochrome-b, and an ATPase. That's it. There are no other protein genes in human mtDNA. All other "mitochondrial proteins" are encoded somewhere else. (That includes 37 out of 44 subunits of the NADH dehydrogenase complex; the DNA polymerase that replicates mitochondrial DNA; the mitochondrial RNA polymerase; about 50 ribosomal proteins; so-called "mitochondrial" catalase; and hundreds of other "mitochondrial" proteins. All are encoded in the nucleus.)
Bottom line: Mitochondrial DNA encodes a very small ensemble of highly specialized membrane-associated proteins. We shouldn't expect this small ensemble to be representative of other genes found in other genomes. (And it's not.) That, in a nutshell, is why mtDNA is not particularly purine-rich in message strands.
But we should test this hypothesis, if possible. (And it is, in fact, possible.) Most bacteria are aerobic, which means most bacterial species have genes for cytochrome-c oxidase, NADH dehydrogenase, etc. The DNA for those genes should be similar to mtDNA with respect to strand-asymmetric purine content. If we analyze bacterial DNA, we should find that genes for cytochrome-c oxidase, NADH dehydrogenase, etc. are pyrimidine-rich on the message strand, just as in mtDNA.
In tomorrow's post: the data.
reade more...
Mitochondrial genes tend to be pyrmidine-rich on the message strand, seemingly in violation of the finding that in all other domains, message strands are purine-rich. The mitochondrial anomaly is actually very easy to understand (although it took me weeks to realize the explanation). In a nutshell: Mitochondrial DNA is pyrimidine-rich on message strands because mtDNA encodes only a few proteins (13, usually), all of them membrane-associated. Membrane-associated proteins are unusual because they tend to incorporate mostly non-polar amino acids such as leucine, isoleucine, valine, proline, alanine, or phenylalanine—all of which are specified by pyrimidine-rich codons.
 |
| The mitochondrion. |
It seems to me mitochondrial DNA shouldn't be thought of as a genome, because well over 90% of mitochondrial-associated gene products are encoded by genes in the host nucleus. (In humans, there may be as many as 1500 nuclear-encoded mitochondrial genes.) This point is worth repeating, so let me quote Patrick Chinnery, TRENDS in Genetics (2003) 19:2, 60:
The vast majority of mitochondrial proteins (estimated at >1000) are synthesized in the cytosol from nuclear gene transcripts.
The circular mitochondrial "chromosome" (if it can be called that) is the vestigial remnant of a much larger genome that long ago migrated to the host nucleus, no doubt to avoid oxidative attack. The mitochondrion simply is not a safe place to store DNA. (Would you set up a sperm bank in a rocket-fuel factory?) It's teeming with molecular oxygen, superoxides, peroxides, free protons, and other hazardous materials.
 |
| The human mitochondrial chromosome. |
Human mitochondrial DNA (which is typical of a lot of mtDNA) encodes just a handful of multi-subnit transmembrane proteins, namely: cytochrome-c oxidase, NADH dehydrogenase, cytochrome-b, and an ATPase. That's it. There are no other protein genes in human mtDNA. All other "mitochondrial proteins" are encoded somewhere else. (That includes 37 out of 44 subunits of the NADH dehydrogenase complex; the DNA polymerase that replicates mitochondrial DNA; the mitochondrial RNA polymerase; about 50 ribosomal proteins; so-called "mitochondrial" catalase; and hundreds of other "mitochondrial" proteins. All are encoded in the nucleus.)
Bottom line: Mitochondrial DNA encodes a very small ensemble of highly specialized membrane-associated proteins. We shouldn't expect this small ensemble to be representative of other genes found in other genomes. (And it's not.) That, in a nutshell, is why mtDNA is not particularly purine-rich in message strands.
But we should test this hypothesis, if possible. (And it is, in fact, possible.) Most bacteria are aerobic, which means most bacterial species have genes for cytochrome-c oxidase, NADH dehydrogenase, etc. The DNA for those genes should be similar to mtDNA with respect to strand-asymmetric purine content. If we analyze bacterial DNA, we should find that genes for cytochrome-c oxidase, NADH dehydrogenase, etc. are pyrimidine-rich on the message strand, just as in mtDNA.
In tomorrow's post: the data.











.png)

.png)
.png)