Point mutations in DNA fall into two types: transition mutations, and transversion mutations. (See graphic below.)

In a transition mutation, a purine is swapped for a different purine (for example, adenine is swapped with guanine, or vice versa), or a pyrimidine is swapped with another pyrimidine (C for T or T for C); and usually, if a purine is swapped on one strand, the corresponding pyrimidine gets swapped on the other. Thus, a GC pair gets changed out for an AT pair, or vice versa.
A transversion, on the other hand, occurs when a purine is swapped for a pyrimidine. In a pairwise sense, this means a GC pair becomes a TA pair (for example) or an AT pair gets changed out for CG, or possibly AT for TA, or GC for CG.
Of the two types of mutation, transitions are more common. We also know that, in particular, GC-to-AT transitions are much more common than AT-to-GC transitions, for reasons that are well understood but that I won't discuss here. If you're curious to know what the experimental evidence is for the greater rate of GC-to-AT transitions, see Hall's 1991 Genetica paper (paywall protected, unfortunately) or the non-paywall-protected Y2K J. Bact. paper by Zhao. The latter paper is interesting because it shows that GC-to-AT transitions are more common in stationary-phase cells than exponentially-growing cells, and also, transitions in stationary E. coli are repaired by MutS and MutL gene products. (Overexpression of those two genes results in fewer transitions. Mutation of those two genes results in more transitions.)
An open question in molecular genetics is: What are the relative rates of transitions versus transversions, in natural populations? We know transitions are more common, but by what factor? Questions like this are tricky to answer, for a variety of reasons, and the answers obtained tend to vary quite a bit depending on the organism and methodology used. Van Bers et al. found a transition/transversion ratio (usually symbolized as κ) of 1.7 in Parus major (a bird species). Zhang and Gerstein looked at human DNA pseudogenes and found transitions outnumber transversions "by roughly a factor of two." Setti et al. looked at a variety of bacteria and found that the transition/transversion rate ratio for mutations affecting purines was 2.1 whereas the rate ratio for pyrimidines was 6.6. Tamura and Nei looked at nucleotide substitutions in the control region of mitochondrial DNA in chimps and humans (a region known to evolve rapidly) and found κ to be approximately 15. Yang and Yoder looked at mitochondrial cytochrome b in 28 primate species and found an average κ of 6.4. (In general, κ values tend to be considerably higher for mitochondrial DNA than other types of DNA.)
It's important to note that in all likelihood, no single value of κ will be universally applicable to all genes in all lineages, because evolutionary pressures vary from gene to gene and the rates of transition and transversion are different for different nucleotides (and so codon usage biases come into play). For an introduction to the various considerations involved in trying to estimate κ, I recommend Yang and Nielsen's 2000 paper as well as their 1998 and 1999 papers.
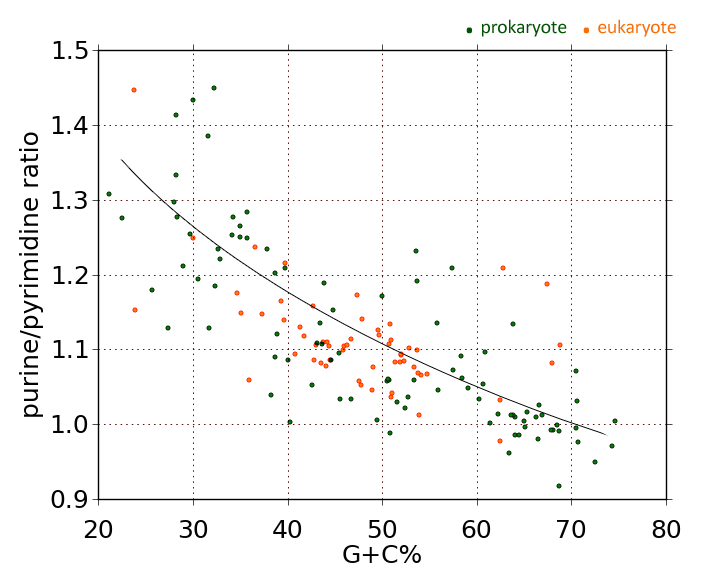
The reason I bring all this up is that I want to offer yet another possible way of estimating the transition/transversion rate ratio κ, using DNA composition statistics. Earlier, I presented data showing that the purine (A+G) content of coding regions of DNA correlates directly with genome A+T content. Analyzing the genomes of representatives of 260 bacterial genera, I came up with the following graph of purine mole-percent versus A+T mole-percent:

The correlation between genome A+T content and mRNA purine content is strong and positive (r=0.852) . Szybalski's Rule says that message regions tend to be purine-rich, but that's not exactly accurate. When genome A+T content is below approximately 35%, coding regions are richer in pyrimidines than purines. Above 35%, purines predominate. The concentration of purines in the mRNA-synonymous strand of DNA rises steadily with genome A+T content. It rises with a slope of 0.13013.
If you try to envision evolution taking an organism from one location on this graph to another, you can imagine that GC-to-AT transitions will move an organism to the right, whereas AT-to-GC transitions will move it to the left. To a first approximation (only!) we can say that horizontal movement on this graph essentially represents the net effect of transitions.
Vertical movement on this graph clearly involves transversions, because a net change in relative A+G content implies nothing less. To a very good first approximation, vertical movement in the graph corresponds to transversions.
Therefore, a good approximation of the relative rate of transitions versus transversions is given by the inverse of the slope. The value comes to 1.0/0.13013, or κ = 7.6846.
In an earlier post, I presented a graph like the one above applicable to mitochondrial DNA (N=203 mitochondrial genomes), which had a slope of 0.06702. Taking the inverse of that slope, we get a value of κ =14.92, which is in excellent agreement with Tamura and Nei's estimate of 15 for mitochondrial κ.
When I made a purine plot using plant and animal virus genomes (N=536), the rise rate (slope) was 0.23707, suggesting a κ value of 4.218. This agrees well with the transition/transversion rate for hepatitus C virus (as measured by Machida et al.) of 1.5 to 7.0 depending on the gene.
In short, we get very reasonable estimates of κ from calculations involving the slope of the A+G vs. A+T graph, across multiple domains.
The main methodological proviso that applies here has to do with the fact that technically, some horizontal movement on the graph can be accomplished with transversions (AT-to-CG, for example). We made a simplifying assumption that all horizontal movement was due to transitions. That assumption is not strictly true (although it is approximately true, since transitions do outnumber transversions; and some transversions, such as AT<-->TA and GC<-->CG, have no effect on genome A+T content). Bottom line, my method of estimating κ probably overestimates κ somewhat, by including a small proportion of AT<-->CG transversions in the numerator. Even so, the estimates agree well with other estimates, tending to validate the general approach.
I invite comments from knowledgeable specialists.
reade more...
In a transition mutation, a purine is swapped for a different purine (for example, adenine is swapped with guanine, or vice versa), or a pyrimidine is swapped with another pyrimidine (C for T or T for C); and usually, if a purine is swapped on one strand, the corresponding pyrimidine gets swapped on the other. Thus, a GC pair gets changed out for an AT pair, or vice versa.
A transversion, on the other hand, occurs when a purine is swapped for a pyrimidine. In a pairwise sense, this means a GC pair becomes a TA pair (for example) or an AT pair gets changed out for CG, or possibly AT for TA, or GC for CG.
Of the two types of mutation, transitions are more common. We also know that, in particular, GC-to-AT transitions are much more common than AT-to-GC transitions, for reasons that are well understood but that I won't discuss here. If you're curious to know what the experimental evidence is for the greater rate of GC-to-AT transitions, see Hall's 1991 Genetica paper (paywall protected, unfortunately) or the non-paywall-protected Y2K J. Bact. paper by Zhao. The latter paper is interesting because it shows that GC-to-AT transitions are more common in stationary-phase cells than exponentially-growing cells, and also, transitions in stationary E. coli are repaired by MutS and MutL gene products. (Overexpression of those two genes results in fewer transitions. Mutation of those two genes results in more transitions.)
An open question in molecular genetics is: What are the relative rates of transitions versus transversions, in natural populations? We know transitions are more common, but by what factor? Questions like this are tricky to answer, for a variety of reasons, and the answers obtained tend to vary quite a bit depending on the organism and methodology used. Van Bers et al. found a transition/transversion ratio (usually symbolized as κ) of 1.7 in Parus major (a bird species). Zhang and Gerstein looked at human DNA pseudogenes and found transitions outnumber transversions "by roughly a factor of two." Setti et al. looked at a variety of bacteria and found that the transition/transversion rate ratio for mutations affecting purines was 2.1 whereas the rate ratio for pyrimidines was 6.6. Tamura and Nei looked at nucleotide substitutions in the control region of mitochondrial DNA in chimps and humans (a region known to evolve rapidly) and found κ to be approximately 15. Yang and Yoder looked at mitochondrial cytochrome b in 28 primate species and found an average κ of 6.4. (In general, κ values tend to be considerably higher for mitochondrial DNA than other types of DNA.)
It's important to note that in all likelihood, no single value of κ will be universally applicable to all genes in all lineages, because evolutionary pressures vary from gene to gene and the rates of transition and transversion are different for different nucleotides (and so codon usage biases come into play). For an introduction to the various considerations involved in trying to estimate κ, I recommend Yang and Nielsen's 2000 paper as well as their 1998 and 1999 papers.
The reason I bring all this up is that I want to offer yet another possible way of estimating the transition/transversion rate ratio κ, using DNA composition statistics. Earlier, I presented data showing that the purine (A+G) content of coding regions of DNA correlates directly with genome A+T content. Analyzing the genomes of representatives of 260 bacterial genera, I came up with the following graph of purine mole-percent versus A+T mole-percent:

The correlation between genome A+T content and mRNA purine content is strong and positive (r=0.852) . Szybalski's Rule says that message regions tend to be purine-rich, but that's not exactly accurate. When genome A+T content is below approximately 35%, coding regions are richer in pyrimidines than purines. Above 35%, purines predominate. The concentration of purines in the mRNA-synonymous strand of DNA rises steadily with genome A+T content. It rises with a slope of 0.13013.
If you try to envision evolution taking an organism from one location on this graph to another, you can imagine that GC-to-AT transitions will move an organism to the right, whereas AT-to-GC transitions will move it to the left. To a first approximation (only!) we can say that horizontal movement on this graph essentially represents the net effect of transitions.
Vertical movement on this graph clearly involves transversions, because a net change in relative A+G content implies nothing less. To a very good first approximation, vertical movement in the graph corresponds to transversions.
Therefore, a good approximation of the relative rate of transitions versus transversions is given by the inverse of the slope. The value comes to 1.0/0.13013, or κ = 7.6846.
In an earlier post, I presented a graph like the one above applicable to mitochondrial DNA (N=203 mitochondrial genomes), which had a slope of 0.06702. Taking the inverse of that slope, we get a value of κ =14.92, which is in excellent agreement with Tamura and Nei's estimate of 15 for mitochondrial κ.
When I made a purine plot using plant and animal virus genomes (N=536), the rise rate (slope) was 0.23707, suggesting a κ value of 4.218. This agrees well with the transition/transversion rate for hepatitus C virus (as measured by Machida et al.) of 1.5 to 7.0 depending on the gene.
In short, we get very reasonable estimates of κ from calculations involving the slope of the A+G vs. A+T graph, across multiple domains.
The main methodological proviso that applies here has to do with the fact that technically, some horizontal movement on the graph can be accomplished with transversions (AT-to-CG, for example). We made a simplifying assumption that all horizontal movement was due to transitions. That assumption is not strictly true (although it is approximately true, since transitions do outnumber transversions; and some transversions, such as AT<-->TA and GC<-->CG, have no effect on genome A+T content). Bottom line, my method of estimating κ probably overestimates κ somewhat, by including a small proportion of AT<-->CG transversions in the numerator. Even so, the estimates agree well with other estimates, tending to validate the general approach.
I invite comments from knowledgeable specialists.

.png)

.png)
.png)