The surprises just keep coming. When you start doing comparative genomics on the desktop (which is so easy with all the great tools at genomevolution.org and elsewhere), it's amazing how quickly you run into things that make you slap yourself on the side of the head and go "Whaaaa????"
If you know anything about DNA (or even if you don't), this one will set you back.
I've written before about Chargaff's second parity rule, which (peculiarly) states that A = T and G = C not just for double-stranded DNA (that's the first parity rule) but for bases in a single strand of DNA. The first parity rule is basic: It's what allows one strand of DNA to be complementary to another. The second parity rule is not so intuitive. Why should the amount of adenine have to equal the amount of thymine (or guanine equal cytosine) in a single strand of DNA? The conventional argument is that nature doesn't play favorites with purines and pyrimidines. There's no reason (in theory) why a single strand of DNA should have an excess of purines over pyrimidines or vice versa, all things being equal.
But it turns out, strand asymmetry vis-a-vis purines and pyrimidines is not only not uncommon, it's the rule. (Some call it Szybalski's rule, in fact.) You can prove it to yourself very easily. If you obtain a codon usage chart for a particular organism, then add the frequencies of occurrence of each base in each codon, you can get the relative abundances of the four bases (A, G, T, C) for the coding regions on which the codon chart was based. Let's take a simple example that requires no calculation: Clostridium botulinum. Just by eyeballing the chart below, you can quickly see that (for C. botulinum) codons using purines A and G are way-more-often used than codons containing pyrimidines T and C. (Note the green-highlighted codons.)

If you do the math, you'll find that in C. botulinum, G and A (combined) outnumber T and C by a factor of 1.41. That's a pretty extreme purine:pyrimidine ratio. (Remember that we're dealing with a single strand of DNA here. Codon frequencies are derived from the so-called "message strand" of DNA in coding regions.)
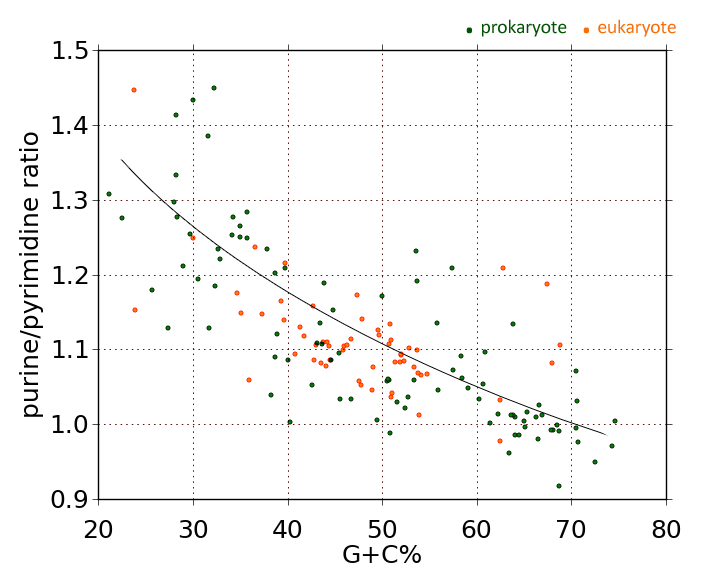
I've done this calculation for 1,373 different bacterial species (don't worry, it's all automated), and the bottom line is, the greater the DNA's A+T content (or, equivalently, the less its G+C content), the greater the purine imbalance. (See this post for a nice graph.)
If you inspect enough codon charts you'll quickly realize that Chargaff's second parity rule never holds true (except now and then by chance). It's a bogus rule, at least in coding regions (DNA that actually gets transcribed in vivo). It may have applicability to pseudogenes or "junk DNA" (but then again, I haven't checked; it may well not apply there either).
If Chargaff's second rule were true, we would expect to find that G = C (and A = T), because that's what the rule says. I went through the codon frequency data for 1,373 different bacterial species and then plotted the ratio of G to C (which Chargaff says should equal 1.0) for each species against the A+T content (which is a kind of phylogenetic signature) for each species. I was shocked by what I found:
I wasn't so much shocked by the fact that Chargaff's rule doesn't hold; I already knew that. What's shocking is that the ratio of G to C goes up as A+T increases, which means G/C is going up even as G+C is going down. (By definition, G+C goes down as A+T goes up.)
Chargaff says G/C should always equal 1.0. In reality, it never does except by chance. What we find is, the less G (or C) the DNA has, the greater the ratio of G to C. To put it differently: At the high-AT end of the phylogenetic scale, cytosine is decreasing faster (much faster) than guanine, as overall G+C content goes down.
When I first plotted this graph, I used a linear regression to get a line that minimizes the sum of squared absolute error. That line turned out to be given by 0.638 + [A+T]. Then I saw that the data looked exponential, not linear. So I refitted the data with a power curve (the red curve shown above) given by
which fit the data even better (minimum summed error 0.1119 instead of 0.1197). What struck me as strange is that the Golden Ratio (1.618) shows up in the power-curve formula (above), but also, the linear form of the regression has G/C equaliing 1.638 when [A+T] goes to 1.0. Which is almost the Golden Ratio.
In a previous post, I mentioned finding that the ratio A/T tends to approximate the Golden Ratio as A+T approaches 1.0. If this were to hold true, it could mean that A/T and G/C both approach the Golden Ratio as A+T approaches 1.0, which would be weird indeed.
For now, I'm not going to make the claim that the Golden Ratio figures into any of this, because it reeks too much of numerology and Intelligent Design (and I'm a fan of neither). I do think it's mildly interesting that A/T and G/C both approach a similar number as A+T approaches unity.
Comments, as usual, are welcome.
reade more...
If you know anything about DNA (or even if you don't), this one will set you back.
I've written before about Chargaff's second parity rule, which (peculiarly) states that A = T and G = C not just for double-stranded DNA (that's the first parity rule) but for bases in a single strand of DNA. The first parity rule is basic: It's what allows one strand of DNA to be complementary to another. The second parity rule is not so intuitive. Why should the amount of adenine have to equal the amount of thymine (or guanine equal cytosine) in a single strand of DNA? The conventional argument is that nature doesn't play favorites with purines and pyrimidines. There's no reason (in theory) why a single strand of DNA should have an excess of purines over pyrimidines or vice versa, all things being equal.
But it turns out, strand asymmetry vis-a-vis purines and pyrimidines is not only not uncommon, it's the rule. (Some call it Szybalski's rule, in fact.) You can prove it to yourself very easily. If you obtain a codon usage chart for a particular organism, then add the frequencies of occurrence of each base in each codon, you can get the relative abundances of the four bases (A, G, T, C) for the coding regions on which the codon chart was based. Let's take a simple example that requires no calculation: Clostridium botulinum. Just by eyeballing the chart below, you can quickly see that (for C. botulinum) codons using purines A and G are way-more-often used than codons containing pyrimidines T and C. (Note the green-highlighted codons.)

If you do the math, you'll find that in C. botulinum, G and A (combined) outnumber T and C by a factor of 1.41. That's a pretty extreme purine:pyrimidine ratio. (Remember that we're dealing with a single strand of DNA here. Codon frequencies are derived from the so-called "message strand" of DNA in coding regions.)
I've done this calculation for 1,373 different bacterial species (don't worry, it's all automated), and the bottom line is, the greater the DNA's A+T content (or, equivalently, the less its G+C content), the greater the purine imbalance. (See this post for a nice graph.)
If you inspect enough codon charts you'll quickly realize that Chargaff's second parity rule never holds true (except now and then by chance). It's a bogus rule, at least in coding regions (DNA that actually gets transcribed in vivo). It may have applicability to pseudogenes or "junk DNA" (but then again, I haven't checked; it may well not apply there either).
If Chargaff's second rule were true, we would expect to find that G = C (and A = T), because that's what the rule says. I went through the codon frequency data for 1,373 different bacterial species and then plotted the ratio of G to C (which Chargaff says should equal 1.0) for each species against the A+T content (which is a kind of phylogenetic signature) for each species. I was shocked by what I found:
 |
| Using base abundances derived from codon frequency data, I calculated G/C for 1,373 bacterial species and plotted it against total A+T content. (Each dot represents a genome for a particular organism.) Chargaff's second parity rule predicts a horizontal line at y=1.0. Clearly, that rule doesn't hold. |
I wasn't so much shocked by the fact that Chargaff's rule doesn't hold; I already knew that. What's shocking is that the ratio of G to C goes up as A+T increases, which means G/C is going up even as G+C is going down. (By definition, G+C goes down as A+T goes up.)
Chargaff says G/C should always equal 1.0. In reality, it never does except by chance. What we find is, the less G (or C) the DNA has, the greater the ratio of G to C. To put it differently: At the high-AT end of the phylogenetic scale, cytosine is decreasing faster (much faster) than guanine, as overall G+C content goes down.
When I first plotted this graph, I used a linear regression to get a line that minimizes the sum of squared absolute error. That line turned out to be given by 0.638 + [A+T]. Then I saw that the data looked exponential, not linear. So I refitted the data with a power curve (the red curve shown above) given by
G/C = 1.0 + 0.587*[A+T] + 1.618*[A+T]2
which fit the data even better (minimum summed error 0.1119 instead of 0.1197). What struck me as strange is that the Golden Ratio (1.618) shows up in the power-curve formula (above), but also, the linear form of the regression has G/C equaliing 1.638 when [A+T] goes to 1.0. Which is almost the Golden Ratio.
In a previous post, I mentioned finding that the ratio A/T tends to approximate the Golden Ratio as A+T approaches 1.0. If this were to hold true, it could mean that A/T and G/C both approach the Golden Ratio as A+T approaches 1.0, which would be weird indeed.
For now, I'm not going to make the claim that the Golden Ratio figures into any of this, because it reeks too much of numerology and Intelligent Design (and I'm a fan of neither). I do think it's mildly interesting that A/T and G/C both approach a similar number as A+T approaches unity.
Comments, as usual, are welcome.



.png)

.png)
.png)